Full HTML
Hemolytic uremic syndrome: An updated review
Elmukhtar Habas1 , Amnna Rayani2 , Ala Habas3 , Kalifa Farfar4 , Eshrak Habas3, Ahmed Elmarghani5, Abdel-Naser Elzouki6
Author Affiliation
1Senior Consultant, Department of Internal Medicine, Hamad General Hospital, Doha, Qatar,
2 Senior Consultant, Department of Pediatrics, Pediatric Hospital, Tripoli, Libya,
3Medical Student, Department of Hematolotugy, Faculty of Medicine, Tripoli University, Tripoli, Libya,
4Consultant Physician, Department of Medicine, Alwakra Hospital, Alwakra, Qatar,
5Assistant Professor, Department of Human Physiology, Biotechnology Research Center, Tripoli, Libya,
6 Senior Consultant, Department of Medicine, Hamad General Hospital, Doha, Qat
Abstract
Hemolytic uremic syndrome (HUS) is a microangiopathic thrombotic disease, which is classified into atypical, typical, and secondary types. Thrombocytopenia, acute kidney failure, and hemolysis are the main features of HUS regardless of its type. Infection with Shiga toxin-producing Escherichia coli causes typical HUS, and gene mutations trigger atypical HUS, while secondary HUS is associated with bone marrow transplantation, autoimmunity, cancer, and other diseases. New insights into the pathogenesis of HUS have emerged over the past decades, suggesting an important role of the complement system in disease pathogenesis, which has been reinforced by the efficacy of plasma exchange and monoclonal antibodies in its treatment. In this review, we performed an updated review of HUS with a focus on understanding its pathogenesis.
DOI: 10.32677/yjm.v1i1.3346
Keywords: Atypical hemolytic uremic syndrome, Hemolysis, Hemolytic uremic syndrome, Renal impairment, Shiga toxin, Thrombocytopenia, Typical hemolytic uremic syndrome
Pages: 6-13
View: 111
Download: 104
DOI URL: https://doi.org/10.32677/yjm.v1i1.3346
Publish Date: 25-03-2025
Full Text
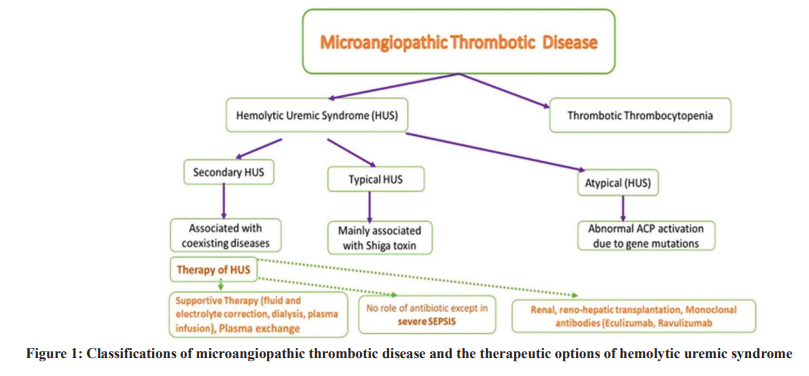
Intravascular hemolysis, microthrombi, low platelet count, and renal impairment are features of thrombotic microangiopathies[1], which, as described by Wardle in 1988, are roughly classified into thrombotic thrombocytopenic purpura (TTP) and hemolytic uremic syndrome (HUS). Despite the similarity in their clinical presentation HUS and TTP have been clearly described as distinct diseases after the identification of von Willebrand factor-cleaving metalloprotease (a disintegrin and metalloprotease with thrombospondin type 1 repeats, member 13 (13ADAMTS13) [2]. HUS is classified into typical, atypical, and secondary subtypes (Fig. 1). The recognized features of HUS occur in HELPP syndrome (elevated hemolysis and liver enzymes and low platelet count) [3], disseminated intravascular coagulation [4], malignant hypertension [5], antiphospholipid antibody syndrome [6], and scleroderma renal crisis [7]. Although there are differences in the pathophysiology of HUS subtypes, clinical differentiation between them is challenging. Typical HUS is the most common type and is associated with Shiga toxins produced by infection with Escherichia coli (E. coli). Atypical HUS is originally due to gene mutations, leading to complement system alternative pathway activation, injuring the vascular endothelial layer. The damaged endothelium causes platelet activation and consumption, resulting in multiple thrombus formations, red blood cell (RBC) lysis, schistocyte, and fragmented RBC formation [8]. Secondary HUS is usually linked to calcineurin inhibitor usage, autoimmune conditions, post-transplantation, viral infection (HIV, Barr, and parvovirus), etc. Before diagnosing atypical HUS, it is essential to rule out primary and secondary HUS, since the treatment options depend on the triggering factors [1]. The aim of this review is to discuss advances in our understanding of the pathogenesis of HUS and the therapeutic options for each subtype.
HUS SUBTYPES
Typical HUS
The worldwide incidence of HUS is 0.5–2.1 cases/106/year, with a peak incidence in children aged <5 years (6.1 cases/106/year). However, there is variation in the incidence of typical HUS in different countries: In the UK, it is 0.91 cases/106/year, in Scotland, 1.25 cases/106 /year, and 1.44 cases/106/year in Canada. It was reported that out of 274 typical HUS cases, 122 cases were children aged <4 years in 2015 in the USA [9].
Typical HUS usually begins after a week of the evident clinical gastroenteritis due to Shiga toxin-producing E. coli infection
(mostly serotype O157:H7 or O104:H4). Clinically, typical HUS patients present with prodromal gastroenteritis (83%), fever (56%), bloody diarrhea (50%) for about 2–7 days, generalized weakness, seizures (20%), acute kidney impairment (97%), anuria (55%), hypertension (47%), severe pallor, and fluid overload (69%). Pathogenesis of typical HUS is largely induced by potent Shiga toxins (Shiga-like toxic protein) that combine with glycolipid Gb3 of the cell membrane in the existence of domain B. The combination causes internalization of domain A which inhibits protein synthesis, increasing the injured cell apoptosis [10]. Furthermore, the E. coli toxins enhance functional tissue factor expression, causing vascular endothelium damage [11]. The damaged endothelium promotes microvascular thrombus creation [12], platelet consumption [8], and RBC lysis [13] (Fig. 2). Gb3 expression is predominant in the glomerular endothelium and in the intestinal endothelial layer. The remarkable Gb3 expression has encouraged scientists to postulate the E. coli gastroenteritis tissue tropism hypothesis. This hypothesis is supported by the simultaneous damage of intestinal and glomerulus endothelium following E. coli infection, especially that produces type II toxin [14].
Atypical HUS (Figs. 1-3)
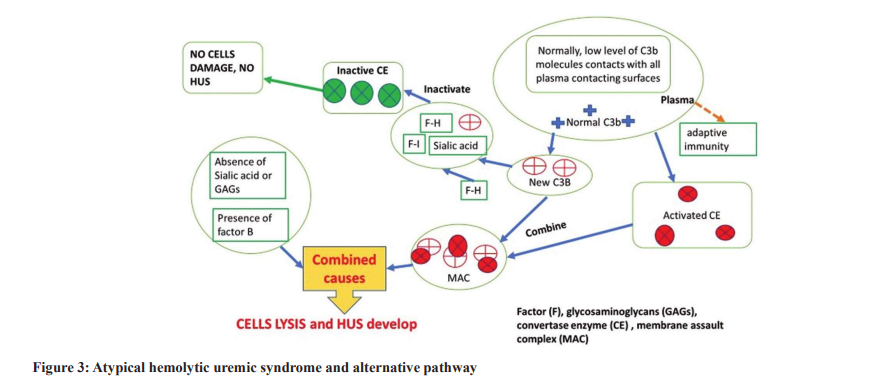
The incidence of atypical HUS is 0.23-0.43 cases/106 /year. The primary underlying cause of the atypical type of HUS is complement dysregulation [15]. It is alleged that the atypical HUS pathogenesis depends upon the disproportionate complement activation by the alternative pathway. The excessive activation of complement is due to built-in regulators, which result from failure to disable C3b deposition on target cell membrane. Complement product deposition injures vessels’ endothelial surface, promoting white blood cell (WBC) and thrombocyte migration and activation, resulting in the creation of thrombi that obstruct blood vessels. The renal impairment that occurs in atypical HUS is primarily due to abnormal complement product depositions in kidney microvasculature, thrombus formation,inducing renal ischemia, and renal impairment [16]. Loss of regulatory protein function is either due to autoantibody formation or genetic mutations. Mutations of some of complement systems factors such as complement factor H (CFH), complement factor I (CFI), membrane cofactor protein (MCP), and complement factor (CF)-3 are found in 40–60% of atypical HUS patients [17], increasing the occurrence rate of atypical HUS. Patients with genetic mutations, infections, autoimmune conditions, drugs, malignancies, or pregnancy are the usual triggers [15,18]. In atypical HUS patients, reported data declared that a mean platelet count usually reduced to 66×103/L [19]. Furthermore, significant renal insufficiency may necessitate dialysis in some cases [19]. TTP cases frequently show either minor or major neurological features; however, atypical HUS could exhibit minor neurologic manifestations [20]. Although the complement C3 product is usually low in serum, C4 is commonly normal; however, they are not diagnostic [21,22]. Atypical HUS due to gene mutations of CFH, CFI, CFB, C3, MCP, CDKE, and THBD CFs can be confirmed by gene sequencing; however, it is not essential to be conducted [22]. Gene studying is expensive, and the results take weeks, delaying the required quick, essentially needed therapy. Approximately half of atypical HUS cases have complement gene mutations or formed autoantibodies [22]. The main differences between typical and atypical HUS pathogeneses began when a link was detected between atypical HUS and the existence of gene mutations, altering complement regulator factor H function in the plasma [23]. Other mutations of complement regulators and CFs such as C3, factor B, factor I, and CD46 were detected recently [24]. A few cases often present rare polymorphism or more than one mutation [25]. The clinical severity of both atypical and typical HUS is almost similar, with a death rate of about 5% [26].
Secondary HUS
Secondary HUS is accompanied by coexisting diseases. Streptococcus pneumoniae and influenza virus infection are the
frequently noted organisms in clinically manifested secondary HUS [27,28]. However, these infections may induce HUS, though they are not triggers of secondary HUS. Differentiation between them to be either trigger or cause is not clear. The pneumococci and influenza A contain active neuraminidase enzymes dedicated to removing sialic acid from cell surface [29,30], causing RBC lysis following complement activation by stimulating alternative pathway [31]
PATHOGENESIS OF HUS
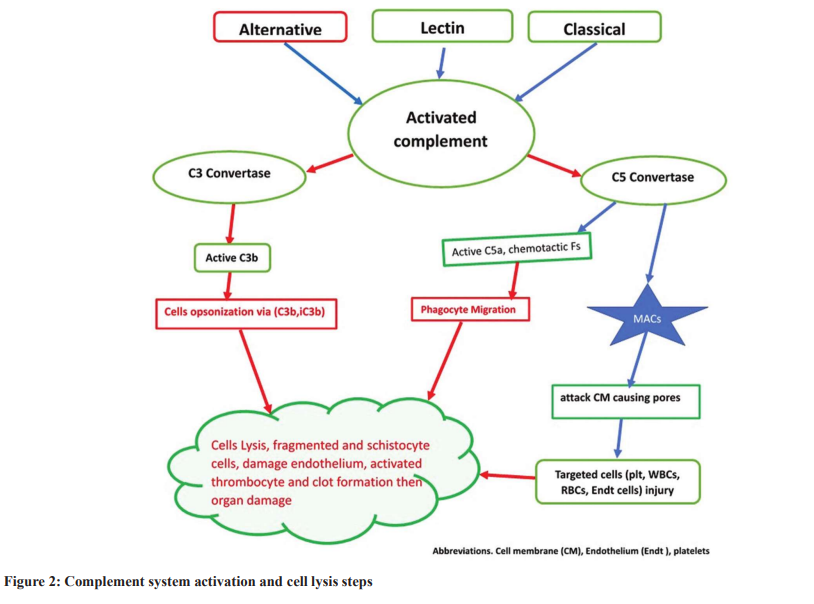
Complement Activation Pathways (Figs. 2 and 3)
Complement activation is a body protective mechanism against infection and disease risks by phagocyte activation or direct lysis. Phagocytosis is enhanced by opsonization in the presence of C3b or its fragments. In addition, phagocyte migration is due to chemotactic and peptides such as C5a synthesis and release (Fig. 2) [16]. Direct target cell lysis by complement via creating membrane pores due to membrane assault complexes (MACs) lysis action (Fig. 3) [32,33]. Besides, it is claimed that complement might initiate and promote adaptive immunity [33]. Complement activation is accomplished via an alternative, lectin, and classical pathways. The alternative path is characterized by persistent low-level C3b on surfaces that have contact with plasma [16]. In abnormal complement activation via the alternative pathway, C3 convertase becomes active, promoting excessive C3b synthesis. The produced C3b initiates cell lysis in the presence of factor B of the cells with no sialic acid glycosaminoglycans on their surface membrane. In addition, the C3 convertase activates the C5 factor (to C5b), promoting WBC sensitization and migration. In addition, C3 convertase promotes MAC formation, initiating cell member pore formation and severe cell damage, direct cell lysis, and cell death [32,33]. Cell opsonization and phagocytosis increase due to C3b and iC3b deposition on the surface of target cells (Fig. 3). In addition, complement initiates and augments adaptive immunity [33].
Endothelial Injury, Leukocyte, and RBC Lysis Mechanism in HUS (Fig. 3)
Complement alternate pathway activation causes deposition of complement assault complex on vascular endothelium cells, RBCs, and leukocytes. Unlike leucocytes and endothelial layer cells, RBCs are probably more prone to fast death due to a lack of cell membrane mending competence, although it is not well documented yet [34]. In atypical HUS, there are two potential mechanisms for RBC destruction, forming RBC fragments and schistocytes [35]. Due to the narrowed vessels, mechanical RBC lysis might occur; however, narrowed capillaries are thought not the cause of RBC lysis or schistocyte formation in HUS in an experimental setting [16]. One of the major weaknesses of this argument is that normal RBCs are usually deformable;
however, RBCs with diameters >3 m are not easily passed through the microvessels with diameters <0.3 m [36]. Secondly,complement-RBCs-mediated lysis is potentially possible;however, the fundamental problem is that the fragmented RBC and schistocyte formation is not a typical feature of RBC destruction in paroxysmal nocturnal hemolytic anemia [37]. Another mechanism involved is possibly due to complement C3d and C3b factor deposition, causing RBC cell membrane stiffness [38]. Therefore, more research is needed to determine what causes the noticeable hemolysis in HUS patients with complement regulatory abnormalities.Blood vessel endothelial cells and WBCs are more efficient than RBC at removing CF deposits and MACs by internalization or scaling [39]. WBC and endothelial cells possibly have a higher ability to repair their damaged membranes than RBCs; however, a histological study of tissues biopsied from patients with acute stage HUS demonstrated endothelial cell destruction [40].Endothelial cell membrane injury and death in cases of typical HUS seem due to the Shiga toxins’ direct effect. In contrast, the damage of the endothelial cells in atypical HUS is assumed due to lytic or sublytic assault by complement [41]. Other mechanisms via nitric oxide, activated neutrophils and monocytes, released reactive oxygen, free hemoglobin, cytokines, procoagulants, and cellular hypoxia may all induce endothelial cell injuries, sharing in atypical HUS pathogenesis.
Complement Activation and Cell Damage in HUS (Figs. 1 and 2)
However, the damaged endothelium may activate the complement cascade [42], and complement activation can happen via any of the three known mechanisms. In atypical HUS, the alternative pathway is the prim activation pathway [43]. Injuries induced by complement activation increase and can cause more damage to surrounding cells, both those in the damaged vicinity and tissues that the complement products pass through [44]. Endothelial cell injury is assumed the initial step in the pathogenesis of both typical and atypical HUS [45]; nevertheless, it is unknown which type of cells is injured first. Complement system activation is further promoted by platelet activation [46] and the released hemoglobin from hemolyzed RBCs [47]. These observations had led to a conclusion that in atypical HUS, the damaged RBCs and the other cells may occur simultaneously or even before endothelial cell damage; however, this is unproven yet [48]. Further studies are encouraged to explore this conclusion.
Platelets Effect in Atypical HUS Pathogenesis
Platelet activation mediated by complement has not been extensively investigated in atypical HUS, such as hemolysis and endothelial cell injury. It appears that platelets may have an essential role in atypical HUS pathophysiology, either directly or indirectly, because of low platelet count [49] and the thrombi containing many platelets [48] are hallmarks of this HUS type. In addition to RBCs, platelets are easily prompted by complement attack [50] via the creation of MACs on their membrane [51]; however, platelets have the power to internalize or shed the complement MACs [52]. It is reported that cell membrane sialic acids protect platelet injury via factor H from the destructive action of the complement [48]. Factor-H and membrane regulators’ combined action protects thrombocytes against complement effect, but this protection is usually not complete in atypical HUS [16]. New research projects are required in this area to clarify the effect of complement on platelets and thrombus formation.
Coagulation-Associated HUS
causes active C5 formation and release or creation of C5b-9 complex, activating endothelial cells and causing procoagulative tissue factor expression [53]. It is reported that various molecular Hemolytic-uremic syndrome On the contrary, toxin levels diminished significantly following azithromycin, despite that E. coli-O157:H7 type may even remain highly viable. It is not recommended to use plasma exchange in interactions between the activated CFs and coagulation typical HUS management. proteins [54]. However, the physiological significance of these interactions has not been undoubtedly established. The vital initiative appears due to thrombocytes’ activation by MACs [50] or C5a [55]. MACs appear to have a procoagulant impact, mediating platelet prothrombinase rather than causing permanent cell destruction [56,57]. Regardless of the cause of thrombocyte activation, it is widely accepted that activation and aggregation of platelets are intimately ended to coagulation, forming another link between complement activation and coagulation process initiation. Despite the abovementioned possible mechanisms of thrombosis, the real mechanism(s) is (are) not clear yet. Further projects are needed to investigate this topic.
HUS THERAPY (FIG. 1)
Acute HUS management is a collaborative achievement between most medical and surgical specialties, especially the transplant teams. The medical teams usually required in acute HUS therapy are internal medicine, intensive care, nephrology, hematology, and neurology. In the acute stage of HUS, full supportive management is the prim therapy. Supportive therapy includes maintaining good fluid hydration, and normal electrolyte balance is essential. It is also vital to maintain controlled blood pressure values, using renin–angiotensin blockade in typical HUS cases and in those who have chronic kidney disease. In 20–40% of HUS patients, especially those with atypical HUS, neurological manifestations such as seizures occur. To avoid severe trauma and brain damage complications, seizure prevention and medical management of the attacks are essential. Dialysis may be indicated in severe azotemia. Treatment was initially similar for all the HUS types. Although atypical HUS in adult cases is treated ironically by plasma exchange, currently, eculizumab and ravulizumab are approved for adult atypical HUS cases. Plasma exchange principally is the first treatment choice in TTP therapy. In contrast, plasma exchange therapy in cases of atypical HUS is less impressive [15]. Furthermore, when the ADAMTS13 activity is normal as expected in most atypical HUS cases, eculizumab therapy should be tried first, and then plasma exchange is advisable when there is no improvement [18]. Antimicrobial therapy is not indicated, except in severe determined sepsis [58].
Treatment of Typical HUS
There is reasonable evidence that antibiotic coverage is unnecessary unless there is well-documented evidence of sepsis [58]. In an in vitro study, initiating antibiotic coverage may inhibit bacterial toxin synthesis to some extent; however, inhibiting DNA synthesis by some antibiotics such as trimethoprim-sulfamethoxazole and ciprofloxacin may increase Shiga toxin formation [59]. In contrast, antibiotics that target organisms’ cell walls, DNA transcription, or translation such as azithromycin do not enhance toxin formation. On the contrary, toxin levels diminished significantly following azithromycin, despite that E. coli-O157:H7 type may even remain highly viable. It is not recommended to use plasma exchange in typical HUS management. In the acute stage of HUS, therapies such as plasma therapy, intravenous globulin, fibrinolysis, antiplatelet, corticosteroids, and even antioxidants have been demonstrated unsuccessful in controlled clinical trials [60]. It was reported that kidney transplantation is advisable in HUS patients, especially children who have a low incidence of end-stage renal failure recurrence rate (0–10%)
Treatment of Atypical HUS
Eculizumab and ravulizumab are approved monoclonal antibodies to treat atypical HUS cases. They prevent complement-mediated microangiopathic thrombus formation. However, it must be emphasized that these two agents are not recommended in patients who had no meningococcal vaccine at least 2 weeks prior to their administration, especially ravulizumab. Plasma exchange must be considered early in atypical HUS therapy (within the earliest 24 h of presentation) and should be continued at least 2 days after complete remission. One or two plasma exchange sessions must be conducted to remove toxins quickly because it reduces mortality significantly. Although plasma infusion may be beneficial in heart or renal failure cases, plasma exchange is preferable because they are more efficient and effective in removing toxins [61]. Intravenous plasma therapy is not indicated HUS induced by S. pneumoniae, while it can aggravate the disease due to the presence of Thomsen–Friedenreich antigen, which stimulates antibody formation by the adults’ plasma. Only one case report has described a reasonable response to a high plasma infusion dose (30 mL/kg/week) over 2 years and a half; however, long-term responses are still under evaluation [60]
Treatment of Secondary HUS
Supportive treatment and controlling the coexisting diseases are the mainstay of this type of HUS. Plasma infusion and plasma exchange can be effective, although they are not commonly conducted.
Eculizumab
Eculizumab is a monoclonal antibody that inhibits C5b-9 synthesis to suppress complement terminal component activation. Eculizumab therapy improved kidney parameters, increased platelets, and reduced the need for hemodialysis in cases that had not responded to plasma therapies. A prospective phase II study reported that non-Shiga toxin-induced HUS cases aged ≥12 years noted a significant improvement in glomerular filtration rate (GFR) following early eculizumab administration. Furthermore, it was reported that the insufficient response to plasma infusion or exchange in patients who improved following eculizumab had supported its effectiveness in treating atypical HUS cases [60]. In phase III prospective study, 41 patients with atypical HUS aged ≥18 years, approximately 75% had normal thrombocyte count and serum lactate dehydrogenase and preserved their kidney function. In addition, life quality improvement, transplant protection, and dialysis withdrawal were reported [22]. However, the number of patients treated with eculizumab was not large enough to draw a firm conclusion; it has been reported that eculizumab has no significant effect in some cases of atypical HUS, particularly those with DGKe mutation [62] or cobalamin C deficiency in adults [63]. However, the number of cases treated was little. Withdrawal of eculizumab therapy after a mean of 16.5 months of follow-up in 36 atypical HUS cases increased the relapse rate in female patients and in cases with MCP, CFH, and CFI complement gene mutations [64], but the need for dialysis decreased in the cases with recurrence of the atypical HUS. The same study reported that 11 of the relapsed patients had recovered to the basal renal function after resuming eculizumab. Unfortunately, 2 cases had deterioration of their increased basal kidney function values, and one patient developed chronic renal and became dialysis dependent [64]. Fakhouri et al. (2016) studied eculizumab which has been given to 41 cases with atypical HUS; platelet count normalized in 98% at a median of 8 days. After 26 weeks, the same study reported that 88% of the cases had sustained normal thrombocyte count and lactate dehydrogenase values, and the estimated GFR improved by ≥15 mL/min/1.73 m2 in 54% of eculizumab treated patients [22]
Ravulizumab
Ravulizumab is another approved monoclonal antibody that blocks CF C5 activation, preventing its cleavage to C5a and C5b, hindering terminal complement complex creation, thereby preventing RBC hemolysis. Ravulizumab is approved for about 30 months for atypical HUS therapy. In two studies, ravulizumab improved HUS completely in 54% of adult atypical HUS cases within the first 26 weeks. Ravulizumab normalized platelets, LDH in 54% of adult cases of atypical HUS, and improved serum creatinine by 25%. Furthermore, ravulizumab therapy has improved thrombocytopenia in 84%, hemolysis in 77%, and improved kidney parameters in about 60% of adults [65]. The two studies showed a higher response rate to ravulizumab in children with atypical HUS than in adult cases.
Transplantation
Membrane-bound protein is highly present in the kidney tissue, and kidney transplantation improves renal function, improves the outcome, and improves the local level of membrane-bound protein expression that helps in dysfunction improvement and the level of membrane-bound protein expression. Kidney transplantation is not a treatment option of either atypical or secondary HUS, while the failure rate is >90%, and these HUS-type recurrences happen in 50%. The recurrence rate in atypical HUS ranges between 30% and 100%, and it is higher in cases with HF1 mutations than in ones with no mutation. The liver synthesizes HF1 protein factor; hence, in genetic defect cases of HF1, it was expected that liver transplantation might improve the outcome. This assumption has encouraged two children to have simultaneous renal and hepatic transplantation, but unfortunately, early liver failure has occurred. This has led to conducting this procedure only for cases that require it as a lifesaving measure.
PREVENTION
Informing health authorities about typical HUS as soon as possible is required since it occurs in epidemics, monitoring the probability of index cases, and imposing preventive measures that reduce disease outbreaks. The only available measure to decrease morbidity and mortality is disease prevention. Antibiotics have been proven ineffective in disease prevention and may augment the HUS risk, especially in children who developed E. coli O157:H7 infection. Therefore, antibiotics are not advisable in HUS except in severely septic patients [58].
CONCLUSION
The characteristic features of the types of HUS are synchronized small vessels’ endothelial cell damage, intravascular RBC lysis, and platelet activation that initiates coagulation, causing small thrombi and tissue ischemic. In atypical HUS pathogenesis, abnormal complement activation via alternative pathway is the main underlying mechanism. Despite the recent evolutions in the HUS pathogenesis, some mechanisms are still not understood clearly and need further research projects. HUS diagnosis on clinical features is always challenging due to the lack of characteristic universal diagnostic criteria, and it is a disease of exclusion. However, gene mutation determination is helpful in atypical HUS diagnosis, but they are expensive and time-consuming and may delay the urgently required therapy. Prevention of HUS is the best therapy option, especially in nontypical HUS cases. Supportive management and conjunction work between the whole managing teams are highly recommended. Plasma exchange and monoclonal antibodies are the best for antiFH-positive HUS therapy.
AUTHORS’ CONTRIBUTIONS
Habas E aided in conceptualization, methodology, literature review, manuscript preparation, critical review, and revisions of the manuscript. Rayani A, Habas A, Farfar K, Habas E, Elmarghani A, and Elzouki A aided in the literature review, manuscript writing, critical review, and revisions of the manuscript. All the authors reviewed and approved the final version of the manuscript.
CONFLICT OF INTEREST
All authors have no conflict of interest.
FUNDING
Nil.
References
1.Arnold DM, Patriquin CJ, Nazy I. Thrombotic microangiopathies: A general approach to diagnosis and management. CMAJ 2017;189:E153-9.
2. Lämmle B, Kremer Hovinga JA, et al. Thrombotic thrombocytopenic purpura. J Thromb Haemost 2005;3:1663-75.
3. Tufano A, Coppola A, Maruotti GM, et al. HELLP syndrome and its relation with the antiphospholipid syndrome. Blood Transfus. 2014;12:114-8.
4. Toh CH, Alhamdi Y, Abrams ST. Current pathological and laboratory considerations in the diagnosis of disseminated intravascular coagulation. Ann Lab Med 2016;36:505-12.
5. Mathew RO, Nayer A, Asif A. The endothelium as the common denominator in malignant hypertension and thrombotic microangiopathy. J Am Soc Hypertens 2016;10:352-9.
6. Rodríguez-Pintó I, Espinosa G, Cervera R. Catastrophic APS in the context of other thrombotic microangiopathies. Curr Rheumatol Rep 2015;17:482.
7. Woodworth TG, Suliman YA, Li W, et al. Scleroderma renal crisis and renal involvement in systemic sclerosis. Nat Rev Nephrol 2016;12:678-91.
8. Jokiranta TS. HUS and atypical HUS. Blood 2017;129:2847-56.
9. Adams DA, Thomas KR, Jajosky RA, et al, Nationally Notifiable Infectious Conditions Group. Summary of notifiable infectious diseases and conditionsUnited States, 2015. MMWR Morb Mortal Wkly Rep 2017;64:1-143.
10. Mukhopadhyay S, Linstedt AD. Manganese blocks intracellular trafficking of Shiga toxin and protects against Shiga toxicosis. Science 2012;335:332-5.
11. Orth-Höller D, Würzner R. Role of complement in enterohemorrhagic Escherichia coli-induced hemolytic uremic syndrome. Semin Thromb Hemost 2014;40:503-7.
12. Grabowski EF, Kushak RI, Liu B, et al. Shiga toxin downregulates tissue factor pathway inhibitor, modulating an increase in the expression of functional tissue factor on endothelium. Thromb Res 2013;131:521-8.
13. Karpman D, Manea M, Vaziri-Sani F, et al. Platelet activation in hemolytic uremic syndrome. Semin Thromb Hemost 2006;32:128-45.
14. Arvidsson I, Stahl AL, Hedström MM, et al. Shiga toxin-induced complement-mediated hemolysis and release of complement-coated red blood cell-derived microvesicles in hemolytic uremic syndrome. J Immunol 2015;194:2309-18.
15. Amaral MM, Sacerdoti F, Jancic C, et al. Action of shiga toxin type-2 and subtilase cytotoxin on human microvascular endothelial cells. PLoS One 2013;8:e70431.
16. Raina R, Krishnappa V, Blaha T, et al. Atypical hemolytic-uremic syndrome: An update on pathophysiology, diagnosis, and treatment. Ther Apher Dial 2019;23:4-21.
17. Alhabhbeh A, Fatima Z, Thomas A, et al. Rare presentation of atypical hemolytic uremic syndrome in an adult. Cureus 2021;13:e18184.
18. Sridharan M, Go RS, Willrich MA. Atypical hemolytic uremic syndrome: Review of clinical presentation, diagnosis and management. J Immunol Methods 2018;461:15-22.
19. Cataland SR, Yang S, Wu HM. The use of ADAMTS13 activity, platelet count, and serum creatinine to differentiate acquired thrombotic thrombocytopenic purpura from other thrombotic microangiopathies. Br J Haematol 2012;157:501-3.
20. Cataland SR, Wu HM. Diagnosis and management of complement mediated thrombotic microangiopathies. Blood Rev 2014;28:67-74.
21. Noris M, Caprioli J, Bresin E, et al. Relative role of genetic complement abnormalities in sporadic and familial aHUS and their impact on clinical phenotype. Clin J Am Soc Nephrol 2010;5:1844-59.
22. Fakhouri F, Hourmant M, Campistol JM, et al. Terminal complement inhibitor eculizumab in adult patients with atypical hemolytic uremic syndrome: A single-arm, open-label trial. Am J Kidney Dis 2016;68:84-93.
23. Warwicker P, Goodship TH, Donne RL, et al. Genetic studies into inherited and sporadic hemolytic uremic syndrome. Kidney Int 1998;53:836-44.
24. Nester CM, Barbour T, de Cordoba SR, et al. Atypical aHUS: State of the art. Mol Immunol 2015;67:31-42.
25. de Córdoba SR. Complement genetics and susceptibility to inflammatory disease. Lessons from genotype-phenotype correlations. Immunobiology 2016;221:709-14.
26. Kielstein JT, Beutel G, Fleig S, et al, Collaborators of the DGfN STECHUS Registry. Best supportive care and therapeutic plasma exchange with or without eculizumab in Shiga-toxin-producing E. coli O104:H4 induced haemolytic-uraemic syndrome: An analysis of the German STEC-HUS registry. Nephrol Dial Transplant 2012;27:3807-15.
27. Allen U, Licht C. Pandemic H1N1 influenza A infection and (atypical) HUS--more than just another trigger? Pediatr Nephrol 2011;26:3-5.
28. Szilágyi A, Kiss N, Bereczki C, et al. The role of complement in Streptococcus pneumoniae-associated haemolytic uraemic syndrome. Nephrol Dial Transplant 2013;28:2237-45.
29. Berry AM, Lock RA, Paton JC. Cloning and characterization of nanB, a second Streptococcus pneumoniae neuraminidase gene, and purification of the NanB enzyme from recombinant Escherichia coli. J Bacteriol 1996;178:4854-60.
30. Gottschalk A. The influenza virus neuraminidase. Nature 1958;181:377-8.
31. Wehling C, Kirschfink M. Tailored eculizumab regimen for patients with atypical hemolytic uremic syndrome: Requirement for comprehensive complement analysis. J Thromb Haemost 2014;12:1437-9.
32. Bhakdi S, Tranum-Jensen J. C5b-9 assembly: Average binding of one C9 molecule to C5b-8 without poly-C9 formation generates a stable transmembrane pore. J Immunol 1986;136:2999-3005.
33. Podack ER, Tschoop J, Muller-Eberhard HJ. Molecular organization of C9 within the membrane attack complex of complement. Induction of circular C9 polymerization by the C5b-8 assembly. J Exp Med 1982;156:268-82.
34. Iida K, Whitlow MB, Nussenzweig V. Membrane vesiculation protects erythrocytes from destruction by complement. J Immunol 1991;147:2638-42.
35. Laurence J. Atypical hemolytic uremic syndrome (aHUS): Making the diagnosis. Clin Adv Hematol Oncol 2012;10:1-12.
36. Chien S. Red cell deformability and its relevance to blood flow. Annu Rev Physiol 1987;49:177-92.
37. Canalejo K, Riera Cervantes N, Felippo M, et al. Paroxysmal nocturnal haemoglobinuria. Experience over a 10 years period. Int J Lab Hematol 2014;36:213-21.
38. Karnchanaphanurach P, Mirchev R, Ghiran I, et al. C3b deposition on human erythrocytes induces the formation of a membrane skeleton-linked protein complex. J Clin Invest 2009;119:788-801.
39. Moskovich O, Herzog LO, Ehrlich M, et al. Caveolin-1 and dynamin-2 are essential for removal of the complement C5b-9 complex via endocytosis. J Biol Chem 2012;287:19904-15.
40. Sethi S, Fervenza FC. Pathology of renal diseases associated with dysfunction of the alternative pathway of complement: C3 glomerulopathy and atypical hemolytic uremic syndrome (aHUS). Semin Thromb Hemost 2014;40:416-21.
41. Manuelian T, Hellwage J, Meri S, et al. Mutations in factor H reduce binding affinity to C3b and heparin and surface attachment to endothelial cells in hemolytic uremic syndrome. J Clin Invest 2003;111:1181-90.
42. Huber-Lang M, Ignatius A, Brenner RE. Role of complement on broken surfaces after trauma. Adv Exp Med Biol 2015;865:43-55.
43. Kang YH, Tan LA, Carroll MV, et al. Target pattern recognition by complement proteins of the classical and alternative pathways. Adv Exp Med Biol 2009;653:117-28.
44. Gorsuch WB, Chrysanthou E, Schwaeble WJ, Stahl GL. The complement system in ischemia-reperfusion injuries. Immunobiology 2012;217:1026-33.
45. Atkinson JP, Liszewski MK, Richards A, et al. Hemolytic uremic syndrome: An example of insufficient complement regulation on self-tissue. Ann N Y Acad Sci 2005;1056:144-52.
46. Del Conde I, Crúz MA, Zhang H, et al. Platelet activation leads to activation and propagation of the complement system. J Exp Med 2005;201:871-9.
47. Frimat M, Tabarin F, Dimitrov JD, et al. Complement activation by heme as a secondary hit for atypical hemolytic uremic syndrome. Blood 2013;122:282-92.
48. Hyvärinen S, Meri S, Jokiranta TS. Disturbed sialic acid recognition on endothelial cells and platelets in complement attack causes atypical hemolytic uremic syndrome. Blood 2016;127:2701-10.
49. George JN, Charania RS. Evaluation of patients with microangiopathic hemolytic anemia and thrombocytopenia. Semin Thromb Hemost 2013;39:153-60.
50. Atefi G, Aisiku O, Shapiro N, et al. Complement activation in trauma patients alters platelet function. Shock 2016;46:83-8.
51. Martel C, Cointe S, Maurice P, et al. Requirements for membrane attack complex formation and anaphylatoxins binding to collagen-activated platelets. PLoS One 2011;6:e18812.
52. Stahl AL, Sartz L, Karpman D. Complement activation on platelet-leukocyte complexes and microparticles in enterohemorrhagic Escherichia coliinduced hemolytic uremic syndrome. Blood 2011;117:5503-13.
53. Tedesco F, Pausa M, Nardon E, et al. The cytolytically inactive terminal complement complex activates endothelial cells to express adhesion molecules and tissue factor procoagulant activity. J Exp Med 1997;185:1619-27.
54. Oikonomopoulou K, Ricklin D, Ward PA, et al. Interactions between coagulation and complement--their role in inflammation. Semin Immunopathol 2012;34:151-65.
55. Rinder CS, Rinder HM, Smith B, et al. Blockade of C5a and C5b-9 generation inhibits leukocyte and platelet activation during extracorporeal circulation. J Clin Invest 1995;96:1564-72.
56. Wiedmer T, Esmon CT, Sims PJ. Complement proteins C5b-9 stimulate procoagulant activity through platelet prothrombinase. Blood 1986;68:875-80.
57. Sims PJ, Wiedmer T. Repolarization of the membrane potential of blood platelets after complement damage: Evidence for a Ca++ -dependent exocytotic elimination of C5b-9 pores. Blood 1986;68:556-61.
58. Wong CS, Jelacic S, Habeeb RL, et al. The risk of the hemolytic-uremic syndrome after antibiotic treatment of Escherichia coli O157:H7 infections. N Engl J Med 2000;342:1930-6.
59. McGannon CM, Fuller CA, Weiss AA. Different classes of antibiotics differentially influence Shiga toxin production. Antimicrob Agents Chemother 2010;54:3790-8.
60. Michael M, Elliott EJ, Ridley GF, et al. Interventions for haemolytic uraemic syndrome and thrombotic thrombocytopenic purpura. Cochrane Database Syst Rev 2009;2009:CD003595.
61. Johnson S, Taylor CM. What’s new in haemolytic uraemic syndrome? Eur J Pediatr 2008;167:965-71.
62. Wehling C, Amon O, Bommer M, et al. Monitoring of complement activation biomarkers and eculizumab in complement-mediated renal disorders. Clin Exp Immunol 2016;187:304-15.
63. Cornec-Le Gall E, Delmas Y, De Parscau L, et al. Adult-onset eculizumabresistant hemolytic uremic syndrome associated with cobalamin C deficiency. Am J Kidney Dis 2014;63:119-23.
64. Fakhouri F, Fila M, Hummel A, et al. Eculizumab discontinuation in children and adults with atypical hemolytic-uremic syndrome: A prospective multicenter study. Blood 2021;137:2438-49.
65. Ultomiris (ravulizumab) Package Insert. Boston, MA: Alexion Pharmaceuticals, Inc.; 2019.